
Bệnh nhân nữ, 71 tuổi nhập viện ngày 8/3/2021 , lý do nhập viện: yếu tái phát 2 chi dưới.
BN khai bệnh khởi phát lần đầu cách 9 năm (năm 2012): Khởi đầu tê hai chân lên đến rốn, tê kiểu châm châm, yếu dần hai chân (bên trái yếu trước), sau đó khoảng 1 tuần liệt hai chân không cử động được. BN không sốt, không nhìn mờ, không bí tiểu. Điều trị tại BV Chợ Rẫy, được chẩn đoán Viêm Tủy Thị Thần Kinh (?)
Diễn tiến:
Bệnh tái phát 2 lần/năm với triệu chứng tương tự. Sau mỗi đợt tái phát BN tự đi lại được, giảm tê hai chân, sinh hoạt bình thường, BN được điều trị (?)
Năm 2013: Bệnh khởi phát với tê và yếu dần sau đó liệt hai chân, có mất thị lực hai mắt, phục hồi thị lực hoàn toàn sau 2 – 3 ngày điều trị tại BV 115.
Năm 2014: đột ngột tê hai chân lan đến rốn, yếu dần hai chân, không mất thị lực, không bí tiểu. Điều trị tại BV ĐHYD đến nay. Truyền Methylprednisolon định kỳ 1 lần/tháng (liều lượng ?) , bệnh tái phát 1 lần/năm.
Năm 2016: giảm thị lực mắt phải dần và đến nay chỉ còn nhìn thấy bóng bàn tay
Hai tháng trước được cấp toa: Medrol 16mg 2.5 viên / ngày + Azathioprine 50 mg 2 viên/ ngày
Cách NV 4 ngày, đột ngột tê hai chân đến rốn, cảm giác dị cảm. Yếu dần hai chân, lúc đầu đi lại cần người dìu, sau đó chỉ cử động được đầu ngón chân. BN đến khám và nhập viện điều trị tại BV ĐHYD.
Tiền sử bản thân:
Được chẩn đoán: Viêm tủy thị thần kinh năm 2012. Từ năm 2014 đến nay điều trị tại BV ĐHYD truyền Methylprednisolone định kỳ mỗi tháng (?). Hai tháng trước BN được cấp toa: Medrol 16mg 2.5 viên / ngày + Azathioprine 50 mg 2 viên/ ngày
Không có tiền sử bệnh lý ngoại khoa; Chấn thương
Chưa ghi nhận tiền căn dị ứng thuốc
Gia đình: không ai có bệnh lý liên quan
Xã Hội: kinh tế khá, không ghi nhận bất thường về dịch tễ
Thăm khám ngày 10/3/2021 (Ngày thứ 3 sau nhập viện)
BN tỉnh táo, chức năng thần kinh cao cấp bình thường
Dây II: Thị lực mắt Phải: Bóng bàn tay; Mắt trái: Đếm ngón tay chính xác cách mắt # 30 cm
Sức cơ: hai tay 5/5; hai chân 1/5 (cử động được ngón chân)
Phản xạ gân cơ: Gối, gót: (-)
Phản xạ da bụng: (-)
Phản xạ tháp: Babinski: đáp ứng duỗi ngón cái hai bên
Cảm giác nông: Giảm cảm giác sờ nông, cảm giác đau từ D10 trở xuống
Cảm giác sâu: Giảm cảm giác run âm thoa ở hai chân; Định vị ngón chính xác
Tóm tắt
BN liệt 2 chi dưới từ năm 2012 được chẩn đoán viêm tủy thị thần kinh, bịnh tái phát nhiều đợt hồi phục tốt, năm 2013 có đợt giảm thị lực 2 mắt hồi phục. Năm 2016 đến nay thị lực 2 mắt không hồi phục. BN nhập viện vì yếu 2 chân tái phát.
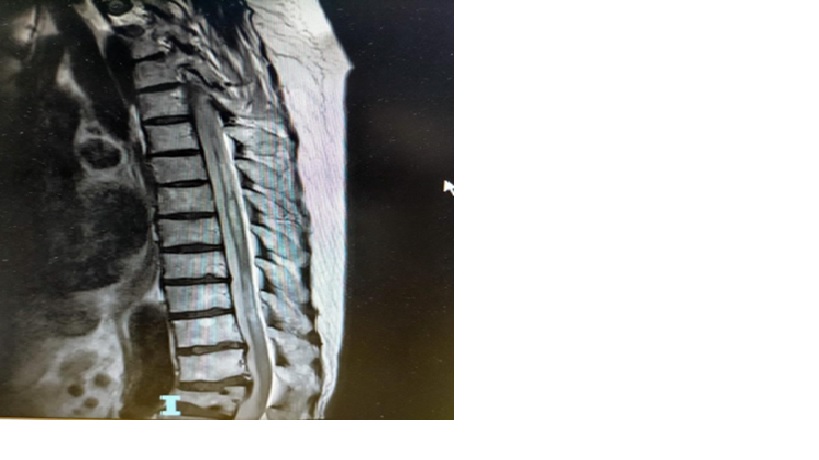
Năm 2016 MRI cột sống (MRI tủy ngực: 1/9/2016)
XN miễn dịch (15/12/2020)
NMO-IgG (anti-AQP4): dương tính
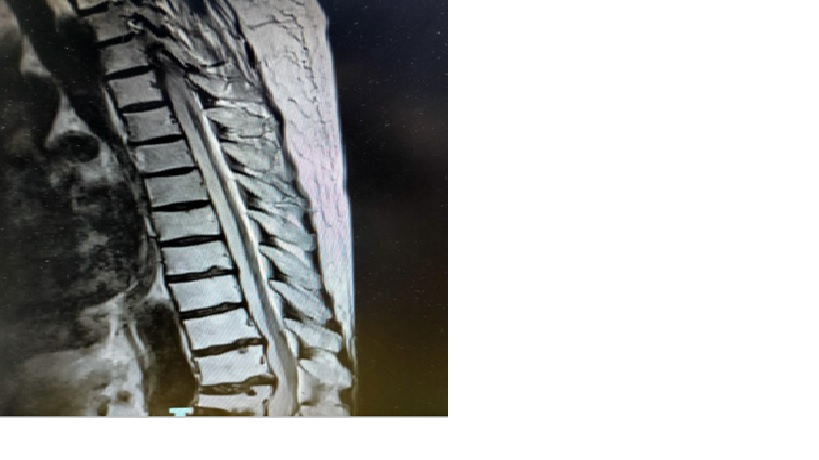
BN được chụp kiểm tra MRI cột sống ngực (8/3/2021)
Teo đoạn T5 – T11 => di chứng do Viêm tủy đã biết. Xẹp thân sống L1
DNT (10/3/2021)
Đường DNT: 7.75 mmol/l / Đường máu: 15.4 mmol/l, Protein: 36.5 mg/dl, Lactate 2.5 mmol/l. Dịch trong, không màu: HC: không, BC: 3 / mm3 (Lympho 100%)
Xét nghiệm thường quy
SH máu: Glucose 220 mg/dl; Ure: 43.74 mg/dl; Creatinin: 0,54 mg/dl; eGFR: 111 ml/ph/1.73 m2; GOT: 15 U/L; GPT: 28 U/L; Na+: 139 mmol/l; K+: 4.02 mmol/l; Ca2+: 2.09 mmol/l;
CRP: 3.6 mmg/l
CTM: HC: 4.47 T/L; Hb: 124 g/l; Hct: 39%; BC: 6.6 G/l ; TC: 154 G/l
Chẩn đoán:
Viêm tủy thị thần kinh tái phát nhiều đợt
Câu hỏi thảo luận
- Bệnh sinh NMO và NMOSD
- Lâm sàng NMO và NMOSD
- Đánh giá và chẩn đoán NMO và NMOSD
- Điều trị và dự hậu
Tổng quan NMO và NMOSD
(Neuromyelitis optica và Neuromyelitis optica spectrum disorders)
Giới thiệu
Neuromyelitis optica (NMO, trước đây được gọi là bệnh Devic) và rối loạn dạng phổ viêm tủy thị thần kinh (neuromyelitis optica spectrum disorder, NMOSD) là những rối loạn viêm của hệ thần kinh trung ương, đặc trưng bởi sự mất myelin nghiêm trọng, qua trung gian miễn dịch và tổn thương sợi trục chủ yếu nhắm vào dây thần kinh thị giác và tủy sống. Kinh điển được coi là một biến thể của bệnh xơ cứng rải rác (MS), NMO hiện được công nhận là một thực thể lâm sàng riêng biệt dựa trên các đặc điểm miễn dịch học . Việc phát hiện ra kháng thể NMO-IgG huyết thanh đặc hiệu cho bệnh liên kết chọn lọc với aquaporin-4 (AQP4) đã giúp tăng cường hiểu biết về các phổ rối loạn khác nhau
Bệnh sinh
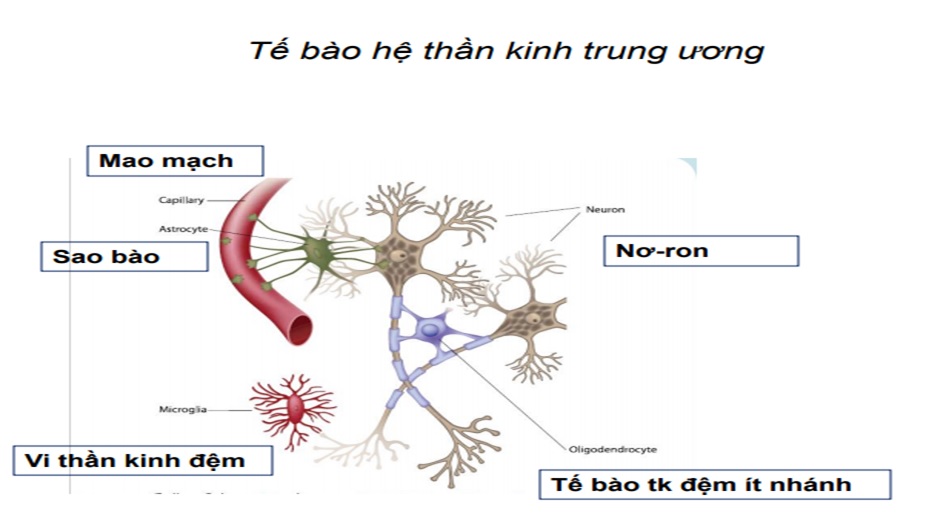
Vai trò sao bào trong hệ thần kinh trung ương
Sao bào (Astrocytes) tìm thấy trong tất cả hệ tktư, có vị trí và vai trò đặc biệt trong BBB, giúp duy trì BBB, sao bào có vai trò sắp xếp quan trọng trong não. Chúng làm việc với tb khác giúp làm mới và hàn gắn myelin trong hệ tktư.
Astrocytes cũng góp phần vào dòng chảy của nước trong hệ tktư. Để làm việc này, astrocytes có nhiều lỗ trống trên bề mặt cho nước vào trong tế bào và được biết như: “aquaporin-4 (AQP4) water channel. Trong MNO, kháng thể trong máu tấn công AQP4 trên astrocytes và ảnh hưởng chức năng của nó
Hàng rào máu não (Blood-brain barrier, BBB)
BBB tách riêng máu và hệ tktư. Hàng rào tồn tại trong tất cả mạch máu rất nhỏ (mao mạch) trong hệ tktư. BBB ngăn ngừa chất không mong muốn vào hệ thần kinh, chỉ cho chất như oxy, glucose, nước đi qua. Trong vài trường hợp bất thường, BBB rò rỉ vài nơi và ảnh hưởng chức năng, một số chất như một vài tế bào máu vào hệ tktư.
Nguyên nhân của NMO và NMOSD vẫn chưa được biết rõ. Cũng như trong bệnh MS, một đợt viêm tự miễn dẫn đến mất myelin và tổn thương sợi trục thông qua các con đường khác nhau.
Trong NMO, quá trình mất myelin và viêm có liên quan đến nhiều đoạn tủy sống và dây thần kinh thị giác liên hệ với mất sợi trục, thâm nhiễm tế bào lympho quanh mạch máu và tăng sinh mạch máu. Không giống như bệnh MS, hoại tử và tạo ra lỗ trống liên quan đến cả chất xám và chất trắng.
Trong khi MS hầu hết là một rối loạn qua trung gian tế bào, sinh lý bệnh của NMO được cho là chủ yếu qua trung gian của hệ thống miễn dịch dịch thể. Một số bằng chứng ủng hộ cơ chế bệnh sinh tự miễn dịch đối với NMO. Điều quan trọng nhất trong số này là việc xác định tự kháng thể đặc hiệu cho bệnh NMO, kháng thể NMO-IgG, còn được gọi là tự kháng thể aquaporin-4 (AQP4).
Hiệu giá tự kháng thể AQP4 huyết thanh tại điểm bắt đầu các cơn lâm sàng đã được chứng minh là tương quan với độ dài tổn thương tủy sống theo chiều dọc. Ngoài ra, hiệu giá kháng AQP4 huyết thanh đã được chứng minh trong một số nghiên cứu liên quan với hoạt động của bệnh, giảm sau khi điều trị ức chế miễn dịch và duy trì ở mức thấp trong thời gian thuyên giảm.
Aquaporin-4 (AQP4), kháng nguyên đích của NMO-IgG, là một protein kênh nước tập trung nhiều ở chất xám tủy sống, các vùng quanh cống não (periaqueductal) và quanh não thất, và sao bào ở hàng rào máu não. Hiện nay NMO-IgG (anti-AQP4) đóng một vai trò trực tiếp trong cơ chế bệnh sinh của NMO.
Dữ liệu bổ sung hỗ trợ cơ chế bệnh sinh tự miễn dịch cho NMO bao gồm các quan sát sau
NMO thường liên quan đến các rối loạn tự miễn dịch toàn thân. Rối loạn cơ quan cụ thể (Organ-specific disorders ) bao gồm: suy giáp, thiếu máu ác tính, viêm loét đại tràng, bệnh nhược cơ và ban xuất huyết giảm tiểu cầu vô căn. Rối loạn không cơ quan cụ thể (Nonorgan-specific disorders ) bao gồm lupus ban đỏ hệ thống, hội chứng kháng phospholipid và hội chứng Sjögren. Ngoài ra, một số trường hợp NMO có thể liên quan đến ung thư
Các tự kháng thể kháng nhân thường gặp ở những bệnh nhân NMO không có bằng chứng về rối loạn toàn thân. Trong một nhóm thuần tập gồm 78 bệnh nhân NMO, độ nhạy huyết thanh với kháng thể kháng nhân (ANA) và Sjögren’s syndrome A / Sjögren’s syndrome B (SSA / SSB) được tìm thấy lần lượt là 53 và 17%.
Trong các bệnh nhân Nhật Bản, bệnh MS tủy sống-thị giác châu Á (Asian optic-spinal multiple sclerosis), hiện được coi là một trong những NMOSD, có liên quan đến alen HLA-DPB1-0501, trong khi bệnh MS thông thường có liên quan đến HLA-DRB1- 1501 alen. Bệnh nhân dương tính với kháng thể kháng AQP4 có nhiều khả năng mang alen HLA-DPB1 hơn.
Kinh nghiệm lâm sàng cho thấy rằng liệu pháp trao đổi huyết tương và các liệu pháp ức chế miễn dịch có lợi cho việc điều trị và phòng ngừa các cơn NMO cấp tính
Lâm sàng NMO và NMOSD
Các đặc điểm nổi bật của NMO bao gồm các cơn cấp tính của viêm dây thần kinh thị giác hai bên hoặc liên tiếp nhanh chóng (dẫn đến mất thị lực nghiêm trọng) hoặc viêm tủy cắt ngang (thường gây yếu chi, mất cảm giác và rối loạn chức năng bàng quang) với đợt tái phát thường xuyên. Các cuộc tấn công thường xảy ra trong nhiều ngày, với mức độ hồi phục thay đổi trong vài tuần đến vài tháng.
Sự tham gia của hệ thần kinh trung ương ngoài dây thần kinh thị giác và tủy sống được ghi nhận ở bệnh nhân NMO và NMOSD. Các triệu chứng gợi ý khác bao gồm các đợt buồn nôn kháng trị, nôn, nấc cụt, buồn ngủ quá mức vào ban ngày hoặc chứng ngủ rũ (narcolepsy), hội chứng bệnh não sau có hồi phục(reversible posterior leukoencephalopathy syndrome), rối loạn nội tiết thần kinh và co giật (ở trẻ em). Mặc dù không có đặc điểm lâm sàng nào là đặc trưng cho bệnh, nhưng một số có đặc trưng cao.
Viêm thị thần kinh (Optic neuritis)
Có thể do bất kỳ tình trạng viêm nhiễm gây ra hoặc có thể vô căn (inflammation of the optic nerve – can be caused by any inflammatory condition or may be idiopathic). Viêm dây thần kinh thị giác biểu hiện với các mức độ mất thị lực khác nhau và hầu như luôn kết hợp với đau mắt, nặng hơn khi cử động của mắt. Các cuộc tấn công viêm dây thần kinh thị giác riêng lẻ trong NMO không thể phân biệt được với các hội chứng riêng biệt của viêm dây thần kinh thị giác hoặc những hội chứng liên quan đến bệnh MS, mặc dù tình trạng mất thị giác thường nghiêm trọng hơn ở NMO. Trong khi phần lớn các cuộc tấn công viêm dây thần kinh thị giác trong NMO là một bên, viêm dây thần kinh thị giác tuần tự liên tiếp nhanh chóng hoặc viêm dây thần kinh thị giác đồng thời hai bên rất gợi ý NMO.
Viêm tủy cắt ngang (Transverse myelitis)
Viêm tủy ngang là rối loạn chức năng tủy sống phát triển trong nhiều giờ hoặc nhiều ngày mà không có tổn thương cấu trúc tủy sống. NMO thường có biểu hiện của viêm tủy cắt ngang, đặc trưng bởi liệt đối xứng hoặc liệt tứ chi, rối loạn chức năng bàng quang và mất cảm giác dưới mức tổn thương tủy sống. Các triệu chứng kèm theo có thể bao gồm co cứng kịch phát (paroxysmal tonic spams ) ở thân hoặc tứ chi, đau rễ, hoặc dấu hiệu Lhermitte.
Ngược lại, viêm tủy trong bệnh MS có xu hướng không hoàn toàn và không đối xứng. Bệnh nhân NMO thường có mức độ mất myelin tủy sống dài hơn so với bệnh nhân MS, thường liên quan đến ba hoặc nhiều đoạn tủy trên MRI, được gọi là viêm tủy cắt ngang lan theo chiều dọc (LETM, longitudinally extensive transverse myelitis ). Tuy nhiên, một số ít bệnh nhân NMO hoặc NMOSD có mức độ tổn thương tủy sống ngắn hơn
Hội chứng thân não (Brainstem syndromes)
Một số bệnh nhân NMOSD có các triệu chứng thân não do liên quan đến hành tủy. •Đặc biệt ở vùng postrema, hội chứng lâm sàng buồn nôn và nôn hoặc nấc cụt, đôi khi kháng trị , với các tổn thương hành tủy trên MRI xảy ra với tỷ lệ mắc bệnh NMOSD từ 16 đến 43%. Sự liên quan đến thân não có thể dẫn đến suy hô hấp cấp tính do thần kinh và tử vong.
Các rối loạn phổ NMO (NMO spectrum disorders )
Một số rối loạn phổ NMO được nhận biết, dựa trên các phát hiện lâm sàng, hình ảnh và kháng thể bao gồm
Các dạng NMO hạn chế hoặc một phần (Limited or partial forms of NMO ):
– Các đợt viêm tủy đơn độc hoặc tái phát, thường nhưng không phải lúc nào cũng liên quan đến tổn thương tủy sống theo chiều dọc (tổn thương tủy sống trên MRI liên quan đến > 3 đoạn đốt sống)
– Viêm dây thần kinh thị giác một bên hoặc hai bên đồng thời, đơn độc hay tái phát
– Viêm dây thần kinh thị giác hoặc viêm tủy cắt ngang riêng biệt
Bệnh MS tủy sống-thị giác Châu Á (Asian optic-spinal multiple sclerosis)
Viêm dây thần kinh thị giác hoặc tổn thương tủy sống lan rộng theo chiều dọc liên quan đến bệnh tự miễn hệ thống
Viêm dây thần kinh thị giác hoặc viêm tủy liên quan đến các tổn thương MRI não riêng biệt điển hình của NMO (ie, with hypothalamic, corpus callosal, periventricular, or periependymal brainstem lesions on T2 images)
Ngoài đặc điểm liên quan đến hệ thần kinh trung ương của NMOSD. Cơ có thể là mục tiêu tấn công trong một số trường hợp hiếm hoi. Có ít nhất một trường hợp báo cáo về một bệnh nhân NMO bị đau cơ tái phát và bằng chứng về một bệnh cơ tự miễn. Ngoài ra, có một số báo cáo về sự tăng cao nhất thời creatine kinase huyết thanh (tức là “tăng CK máu”) liên quan đến các cuộc tấn công của NMOSD.
Theo thời gian, danh mục rối loạn phổ NMO đã mở rộng bao gồm những bệnh nhân có tình trạng dương tính với kháng thể AQP4 bị các đợt tấn công đơn độc hoặc tái phát của viêm dây thần kinh thị giác, viêm tủy, hội chứng thân não hoặc hội chứng não, thường không thể phân biệt được với bệnh MS
Các biểu hiện khác
Các biểu hiện khác có thể phát triển với NMO và NMOSD bao gồm bệnh não, fulminant cerebral demyelination, rối loạn chức năng vùng dưới đồi và bệnh não sau có hồi phục .
Các triệu chứng liên quan đến tổn thương vùng dưới đồi hai bên có thể bao gồm triệu chứng ngủ rũ hoặc buồn ngủ quá mức vào ban ngày, béo phì và các biểu hiện TK tự động khác nhau như hạ huyết áp, nhịp tim chậm và hạ thân nhiệt . Trong một số trường hợp hiếm hoi, phù mạch lan tỏa tối cấp (fulminant diffuse vasogenic edema) có thể dẫn đến thoát vị não và tử vong .
Đau là một triệu chứng phổ biến với NMO. Trong các nghiên cứu hồi cứu về NMO và NMOSD, 80 phần trăm bệnh nhân trở lên báo cáo đau, thường liên quan đến thân và chân
Tiến triển của bệnh
NMO có một đợt tái phát trong 90 phần trăm trường hợp trở lên. Ở một số bệnh nhân, viêm dây thần kinh thị giác và viêm tủy cắt ngang xảy ra đồng thời; ở những người khác, các giai đoạn lâm sàng được phân tách bởi thời gian trì hoãn thay đổi.
Tái phát xảy ra trong năm đầu tiên sau một biến cố ban đầu ở 60 phần trăm bệnh nhân và trong vòng ba năm ở 90 phần trăm. Theo quy luật, thiếu hụt còn lại nghiêm trọng theo sau các cuộc tấn công ban đầu và sau đó, dẫn đến sự phát triển nhanh chóng của tàn tật do mù lòa và liệt 2 chi trong vòng năm năm.
Không giống như MS, giai đoạn tiến triển thứ phát của bệnh rất hiếm. Bệnh nhân có biểu hiện não có thể bị các biểu hiện ở não tiếp tục bị ở não mà không có sự tham gia của các dây thần kinh thị giác hoặc tủy sống.
Đánh giá và chẩn đoán
Ngoài tiền sử và thăm khám toàn diện, việc đánh giá nghi ngờ NMOSD còn bao gồm việc kiểm tra não và tủy sống bằng MRI, xác định tự kháng thể trong huyết thanh aquaporin-4 (AQP4) IgG và phân tích dịch não tủy. Việc phát hiện kháng thể IgG AQP4 là đặc hiệu để xác nhận chẩn đoán trong các cơ sở lâm sàng thích hợp.
Các biểu hiện lâm sàng nghi ngờ NMOSD bao gồm những điều sau đây
Viêm dây thần kinh thị giác đồng thời xảy ra hai bên, liên quan đến giao thoa thị giác, (Optic neuritis that is simultaneously bilateral, involves the optic chiasm, causes an altitudinal visual field defect, or causes severe residual visual loss).
Viêm tủy sống cắt ngang hoàn toàn (nhiều hơn không hoàn toàn), đặc biệt với các cơn co thắt kịch phát (A complete (rather than partial) spinal cord syndrome, especially with paroxysmal tonic spasms).
Hội chứng lâm sàng vùng postrema bao gồm nấc cụt khó chữa hoặc buồn nôn và nôn (An area postrema clinical syndrome consisting of intractable hiccups or nausea and vomiting)
Tuy nhiên, không có biểu hiện nào trong số này là chẩn đoán NMOSD khi không phát hiện được kháng thể AQP4 IgG và ngược lại, dạng phổ NMO có thể rộng hơn, dựa trên sự hiện diện của kháng thể AQP4 IgG trong các hội chứng tủy sống nhẹ hơn.
Tiêu chuẩn chẩn đoán
Tiêu chí đồng thuận sửa đổi được công bố năm 2015 thống nhất các khái niệm về NMO và NMOSD và chẩn đoán dựa trên sự hiện diện của các đặc điểm lâm sàng cốt lõi, tình trạng kháng thể AQP4 và các đặc điểm hình ảnh thần kinh MRI. Các tiêu chí công nhận sáu đặc điểm lâm sàng cốt lõi
Kháng thể NMO dương tính

Kháng thể NMO âm tính hay không thực hiện được

Thêm tiêu chuẩn MRI

Không điển hình NMOSD

Cận lâm sàng
MRI tủy sống
Tổn thương tủy sống lan rộng theo chiều dọc trên MRI T2W, đặc biệt là những tổn thương kéo dài từ ba đoạn đốt sống trở lên và chủ yếu liên quan đến chất xám của tủy sống trung tâm trên các phần trục, gợi ý nhiều đến NMO. Các tổn thương cấp tính thường liên quan đến hầu hết diện tích mặt cắt ngang của một đoạn tủy sống, với hiện tượng phù nề và tăng sinh gadolinium. Dấu hiệu “mắt cú” (“owl-eye” ) là do tế bào sừng trước trong chất xám tủy sống tăng đậm độ, gợi ý thiếu máu cục bộ động mạch tủy sống.
Tủy cổ bị ảnh hưởng trong khoảng 60% trường hợp, và các tổn thương có thể kéo dài vào hành tủy. Đôi khi, tình trạng viêm và sung phù tủy sống nghiêm trọng đến mức tổn thương có thể giống một khối u. Sự tăng cường Gadolinium biến mất khi điều trị và các tổn thương tủy sống giảm dần trong quá trình thuyên giảm.
MRI não và thần kinh thị giác
MRI não là bình thường ở 55 đến 84 phần trăm bệnh nhân NMO, ngoài việc tăng cường gadolinium của các dây thần kinh thị giác. Tuy nhiên, theo thời gian, bằng chứng MRI về sự liên quan của não phát triển ở 85% bệnh nhân mắc NMO. Các tổn thương được mô tả ở trung tâm hành tủy, vùng dưới đồi và màng não, tương ứng với các vùng biểu hiện AQP4 cao, nhưng cũng được tìm thấy trong chất trắng dưới vỏ. Những tổn thương này ở những bệnh nhân NMO hoặc NMOSD đôi khi đáp ứng các tiêu chuẩn chẩn đoán MS về sự lan tỏa trong không gian.
Tự kháng thể AQP4
Tự kháng thể huyết thanh aquaporin-4 (AQP4), còn được gọi là NMO-IgG, là một dấu ấn sinh học cụ thể cho NMOSD. Thụ thể aquaporin-4 là kháng nguyên đích của NMO-IgG, có vai trò trực tiếp trong quá trình sinh bệnh của NMO. Do đó, những bệnh nhân nghi ngờ mắc NMO cần được xét nghiệm tìm kháng thể IgG AQP4 trong huyết thanh. Tốt nhất, xét nghiệm kháng thể AQP4 nên được thực hiện trong khi tấn công và trước khi điều trị ức chế miễn dịch. Ngoài ra, những bệnh nhân ban đầu có huyết thanh âm tính với kháng thể AQP4 nên được xét nghiệm lại nếu nghi ngờ NMO.
Xét nghiệm huyết thanh NMO-IgG ban đầu cho thấy độ nhạy vừa phải và độ đặc hiệu cao để phát hiện NMO (tương ứng là 73 và 91%). Ngay cả khi sử dụng các xét nghiệm nhạy cảm nhất, 12% bệnh nhân được chẩn đoán lâm sàng về NMO hoặc NMOSD đều có huyết thanh âm tính với NMO-IgG. Dữ liệu hạn chế gợi ý rằng NMOSD huyết thanh âm tính có thể khác với NMOSD huyết thanh dương tính ở một số đặc điểm nhất định, bao gồm tỷ lệ nam nữ bằng nhau, chủ yếu là dân tộc da trắng và nhiều khả năng bị đồng thời viêm dây thần kinh thị giác và viêm tủy ngang ở lần xuất hiện đầu tiên.
Tự kháng thể MOG
Một số ít bệnh nhân có kháng thể chống lại myelin oligodendrocyte glycoprotein (MOG), nhưng mức độ liên quan lâm sàng của kháng thể kháng MOG trong NMOSD là không chắc chắn. Các tự kháng thể MOG có thể xác định một hội chứng lâm sàng chồng chéo thường đáp ứng các tiêu chí lâm sàng cho NMOSD nhưng có một số khác biệt về các tính năng so với NMOSD liên kết với kháng thể AQP4 hoặc NMOSD âm tính, bao gồm:
Tỷ lệ nam giới bị ảnh hưởng nhiều hơn
Có nhiều khả năng liên quan đến dây thần kinh thị giác hơn tủy sống
Viêm dây thần kinh thị giác hai bên đồng thời thường xuyên hơn
Có nhiều khả năng là một pha
Tổn thương tủy sống chủ yếu xảy ra ở phần dưới của tủy sống
Dịch não tủy
Trong các đợt cấp tính của NMO, bất thường về dịch não tủy (CSF) thường gặp, bao gồm tăng bạch cầu và tăng nồng độ protein (pleocytosis and elevated protein levels). Tăng bạch cầu được phát hiện ở 14 đến 79 phần trăm bệnh nhân bị NMO, điển hình là bạch cầu đơn nhân hoặc tế bào lympho, mặc dù bạch cầu trung tính có thể chiếm ưu thế. Số lượng bạch cầu CSF > 50 tế bào / mm3 được báo cáo ở 13 đến 35 phần trăm bệnh nhân NMO; những người bị tổn thương tủy sống theo chiều dọc tỷ lệ cao hơn so với những người bị viêm dây thần kinh thị giác. Đáng chú ý, oligoclonal bands thường không có (70 đến 85 phần trăm các trường hợp). Ngược lại, tỷ lệ bạch cầu trong dịch não tủy> 50 tế bào / mm3 hiếm gặp trong MS trong khi oligoclonal bands có ở hơn 90% bệnh nhân.
Các nghiên cứu chụp cắt lớp kết hợp quang học trong NMO báo cáo rằng lớp sợi thần kinh võng mạc mỏng hơn đáng kể ở bệnh nhân NMO so với bệnh MS, phản ánh sự tổn thương sợi trục nghiêm trọng hơn. Microcystic macular edema of the inner nuclear dường như phổ biến ở những bệnh nhân bị NMO và có tiền sử viêm dây thần kinh thị giác. Tuy nhiên, tiện ích của chụp cắt lớp kết hợp quang học như một công cụ chẩn đoán chưa được thiết lập tốt.
Chẩn đoán phân biệt NMO và NMOSD
Các hội chứng NMO phải được phân biệt với bệnh MS, rối loạn phổ biến nhất trong mất myelin của hệ thần kinh trung ương. Viêm não lan tỏa cấp tính và các bệnh tự miễn khác như lupus ban đỏ hệ thống và bệnh Behçet hiếm khi có các biểu hiện tương tự.
Cần lưu ý, tổn thương tủy sống lan rộng theo chiều dọc(LETM) không đặc hiệu cho NMO. Gặp ở bệnh nhân mắc các bệnh tự miễn dịch hoặc viêm nhiễm khác, bao gồm lupus ban đỏ hệ thống, hội chứng Sjögren, bệnh thần kinh-Behçet, bệnh sarcoidosis, bệnh MS, rối loạn cận nhiễm (parainfectious disorders) (ví dụ, viêm não lan tỏa cấp tính) và viêm não kháng thụ thể NMDA . Các nguyên nhân bổ sung của tổn thương tủy sống lan rộng theo chiều dọc bao gồm khối u nội tủy, nguyên nhân mạch máu (ví dụ, lỗ rò động mạch màng cứng tủy sống và nhồi máu do tắc động mạch cột sống trước), chuyển hóa (ví dụ, thiếu vitamin B12 gây thoái hóa kết hợp bán cấp của tủy sống) , xạ trị và nhiễm vi-rút (ví dụ: HIV-1, HTLV-1).
Phân biệt NMO với các bệnh mất myelin khác
Việc phân biệt NMO với các bệnh mất khác dựa trên những khác biệt quan trọng về diễn biến lâm sàng, tiên lượng và sinh lý bệnh cơ bản cũng như khả năng đáp ứng với các liệu pháp điều chỉnh bệnh MS. Một số đặc điểm dường như để phân biệt NMO với bệnh MS tái phát-thuyên giảm cổ điển:
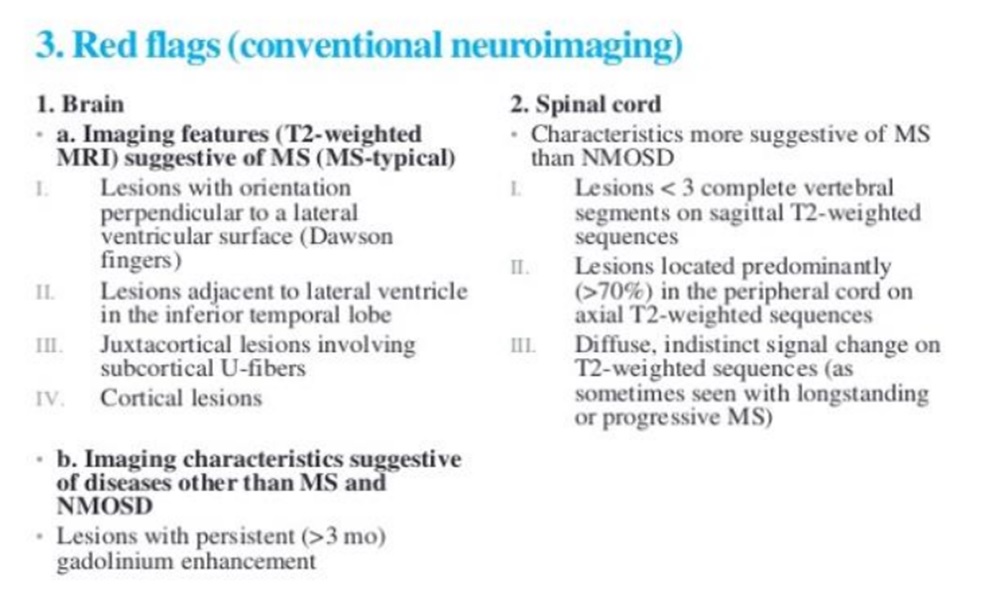
MRI não thường bình thường ở bệnh nhân NMO, đặc biệt khi mới khởi phát, và MRI tủy sống thường biểu hiện các tổn thương rộng trên ba hoặc nhiều đoạn đốt sống. Tuy nhiên, bằng chứng lâm sàng hoặc MRI về sự liên quan của não, đặc biệt là ở thân não, xuất hiện ở một tỷ lệ đáng kể bệnh nhân NMO. Các phát hiện trên MRI não gợi ý chẩn đoán MS hơn là NMO bao gồm các tổn thương T2W ở một hoặc nhiều vị trí sau:
• Tổn thương tiếp giáp với não thất bên (Lesions adjacent to lateral ventricle)
• Tổn thương chất trắng ở thùy thái dương dưới
• Tổn thương hình trứng (tức là “ngón tay Dawson”) quanh não thất
• Tổn thương sợi U cận vỏ (U-fiber juxtacortical lesions)
Tuy nhiên, những phát hiện hình ảnh thần kinh này không nhất thiết loại trừ chẩn đoán NMO, vì chúng có thể xảy ra ở những bệnh nhân NMO có huyết thanh dương tính với kháng thể AQP4.
Trong các đợt cấp của NMO, dịch não tủy (CSF) có thể biểu hiện tăng bạch cầu đa nhân trung tính, nhưng thường (70 đến 85% trường hợp) âm tính với oligoclonal bands. Việc phát hiện dương tính với kháng thể AQP4 là đặc hiệu cho NMO và NMOSD. Bệnh lý tủy và viêm dây thần kinh thị giác liên quan đến NMO có xu hướng nghiêm trọng hơn so với bệnh MS, khả năng hồi phục ít hơn.
Điều trị NMO và NMOSD
Đợt cấp và cơn tái phát (Acute attacks and relapses of NMO)
Các đợt cấp và tái phát của NMO thường được điều trị bằng glucocorticoid tiêm tĩnh mạch, ngay sau đó là thay huyết tương trị liệu đối với các triệu chứng kháng trị hoặc tiến triển. Để phòng ngừa các đợt tái phát, điều trị bằng ức chế miễn dịch toàn thân là phương pháp chính.
(Tuy nhiên, không có thử nghiệm đối chứng nào đánh giá việc điều trị NMO và các khuyến cáo chủ yếu từ dữ liệu các nghiên cứu quan sát và kinh nghiệm lâm sàng của các chuyên gia)
Cơ sở lý luận của việc điều trị các cơn cấp tính và tái phát ở NMO dựa trên bằng chứng cho thấy khả năng tự miễn dịch thể dịch đóng một vai trò trong cơ chế bệnh sinh của NMO, và được thúc đẩy bởi tình trạng tàn tật liên quan đến cơn cao hơn, tiên lượng xấu và nguy cơ tử vong cao ở những bệnh nhân không được điều trị.
Khởi đầu và tiếp theo cơn cấp (Initial and subsequent acute attacks)
Tất cả các bệnh nhân nghi ngờ NMO nên được điều trị các đợt cấp. Điều trị khởi đầu bằng methylprednisolone tiêm tĩnh mạch liều cao (1 gam mỗi ngày trong 3-5 ngày liên tục), phù hợp với khuyến cáo của hội đồng chuyên gia và dựa trên các nghiên cứu về bệnh MS và viêm dây thần kinh thị giác vô căn.
Một số tác giả đề nghị
Methylprednisolone 1g TM trong 5 ngày tiếp theo uống prednisone(1mg/kg cân nặng cơ thể và giảm dần trên 6-12 tháng.
Đối với những bệnh nhân có các triệu chứng nặng, không đáp ứng với glucocorticoid, thay huyết tương là phương pháp điều trị được gợi ý. Dữ liệu hồi cứu hạn chế và không được kiểm chứng cho thấy rằng điều trị ban đầu bằng glucocorticoid đường tĩnh mạch cộng với thay huyết tương kết quả cải thiện so với điều trị bằng glucocorticoid đơn thuần. Thay huyết tương được thực hiện cách ngày với tổng số bảy lần thay. Globulin miễn dịch tiêm tĩnh mạch chưa được đánh giá cụ thể đối với các đợt cấp của NMO và hiếm khi được sử dụng trong điều kiện này.
Mục tiêu điều trị cấp:
- ức chế viêm nhiễm (suppress acute inflammatory attack)
- giảm tổn thương hệ tktư (minimize CNS damage)
- cải thiện chức năng tk lâu dài (improve long-term neurological function)
Phòng ngừa cơn tấn công (Attack prevention)
Khuyến cáo bắt đầu điều trị ức chế miễn dịch dài hạn để phòng ngừa các cuộc tấn công ngay sau khi chẩn đoán NMO được đưa ra. Dữ liệu về hiệu quả của các liệu pháp phòng ngừa được dựa trên các nghiên cứu quan sát.
Nền tảng của điều trị là ức chế miễn dịch toàn thân với các tác nhân bao gồm: azathioprine, mycophenolate mofetil, rituximab, methotrexate, mitoxantrone và glucocorticoid đường uống.
Phòng ngừa tái phát
Mục đích điều trị NMO là giảm và phòng ngừa tái phát
Immunotherapies (steroid: prednisolone)
Thêm vào azathioprine, methotrexate hay mycophenolate cho phép giảm steroids.
Khi điều trị ức chế hệ thống miễn dịch, nguy cơ cao nhiễm trùng, theo dõi công thức máu, chức năng gan, thận
Chế độ thuốc tối ưu và thời gian điều trị vẫn chưa được xác định. Mặc dù không có sự đồng thuận chặt chẽ, các tác nhân thường được coi là phương pháp điều trị đơn trị liệu đầu tay cho NMO là azathioprine, rituximab và mycophenolate mofetil.
Dữ liệu so sánh còn ít ỏi, nhưng một nghiên cứu hồi cứu, không phân loại từ hai trung tâm đại học ở Hoa Kỳ đã phân tích sự tái phát ở những bệnh nhân NMO hoặc NMOSD được điều trị bằng azathioprine và prednisone đồng thời (n = 32) trong ít nhất sáu tháng, hoặc mycophenolate (n = 28) trong ít nhất sáu tháng, hoặc với rituximab (n = 30) trong ít nhất một tháng và theo dõi sau khi điều trị ít nhất sáu tháng. Điều trị bằng cả ba tác nhân có liên quan đến việc giảm đáng kể tỷ lệ tái phát hàng năm, từ 72 đến 88 phần trăm so với ban đầu. Ví dụ, tỷ lệ tái phát hàng năm giảm từ 2,26 trước khi điều trị bằng azathioprine xuống 0,63 sau khi điều trị, giảm 72 phần trăm.
Điều trị thất bại được định nghĩa là sự phát triển của bất kỳ trường hợp viêm nhiễm hệ thần kinh trung ương mới nào xảy ra mặc dù đã điều trị ức chế miễn dịch; tỷ lệ thất bại điều trị với những loại thuốc này thay đổi từ 33 đến 53%.
Thời gian điều trị
Sự ức chế miễn dịch thường được tiếp tục trong ít nhất 5 năm đối với những bệnh nhân có huyết thanh dương tính với AQP4, kể cả những bệnh nhân có biểu hiện của một đợt tấn công đơn lẻ, vì họ có nguy cơ cao tái phát hoặc chuyển sang NMO.
Tuy nhiên, không có sự nhất trí về thời gian điều trị ức chế miễn dịch. Một số chuyên gia gợi ý rằng liệu pháp kéo dài cuộc sống là thích hợp, với bản chất thường là tàn phá của căn bệnh này. Những người khác gợi ý rằng thời gian ức chế miễn dịch nên được điều chỉnh cho phù hợp với mức độ nghiêm trọng của các cuộc tấn công và tình trạng tàn tật.
Vai trò các thuốc mới
Điều trị tocilizumab (chất đối kháng thụ thể IL-6) có liên quan đến việc ổn định hoặc cải thiện lâm sàng ở một số ít bệnh nhân mắc NMO kháng trị đã thất bại một hoặc nhiều phương pháp điều trị “tiêu chuẩn” được thảo luận ở trên.
Tương tự, điều trị NMO bằng eculizumab (kháng thể ức chế bổ thể) có liên quan đến việc giảm đáng kể tần suất tấn công trong một nghiên cứu mở không kiểm chứng nhỏ,
Bằng chứng quan sát hạn chế cho thấy rằng điều trị NMO bằng interferon beta, natalizumab, hoặc fingolimod không hiệu quả và có thể có hại . Không có bằng chứng được công bố liên quan đến việc điều trị NMO bằng ocrelizumab.
Dự hậu
Bệnh sử tự nhiên của NMO là một trong những suy thoái từng bước do tích lũy các khiếm khuyết về thị giác, vận động, giác quan và bàng quang từ các cuộc tấn công tái phát.
Hầu hết các cơn cấp tính hoặc tái phát đều nặng hơn trong nhiều ngày và hồi phục trong vài tuần đến vài tháng với những di chứng đáng kể.
Các yếu tố dự báo tiên lượng xấu hơn bao gồm số lần tái phát trong vòng hai năm đầu, mức độ nghiêm trọng của cơn đầu tiên, tuổi lớn hơn khi khởi phát bệnh, và (có thể) liên quan với các rối loạn tự miễn khác.
Tỷ lệ tử vong cao ở NMO, thường gặp nhất là thứ phát sau suy hô hấp do thần kinh,
Các nghiên cứu thuần tập về các quần thể Tây Ấn, Bắc Mỹ, Brazil và Pháp đã báo cáo tỷ lệ tử vong lần lượt là 32 phần trăm, 50 phần trăm và 25 phần trăm ở NMO. Những nghiên cứu này có thể thiên về những trường hợp nghiêm trọng hơn. Tiến bộ trong chẩn đoán và điều trị NMO dự kiến sẽ làm giảm tỷ lệ tử vong.
Tự kháng thể AQP4 (NMO-IgG) có thể là dấu hiệu cho diễn biến bệnh và tiên lượng, mặc dù dữ liệu hiện có không nhất quán. Ở những bệnh nhân bị viêm dây thần kinh thị giác tái phát, bằng chứng hồi cứu cho thấy NMO-IgG có liên quan đến kết quả thị giác kém và sự phát triển của NMO.
Một nghiên cứu tiền cứu trên 29 bệnh nhân có biểu hiện tổn thương tủy sống lan rộng theo chiều dọc cho thấy 55% bệnh nhân có huyết thanh dương tính với NMO-IgG tái phát trong vòng một năm hoặc chuyển sang NMO, trong khi không có bệnh nhân âm tính nào tái phát. Ngược lại, một báo cáo tiếp theo lưu ý rằng NMO âm tính và huyết thanh dương tính tương tự nhau về tỷ lệ tái phát, mức độ nghiêm trọng và kết cục lâu dài. Chỉ có dữ liệu hạn chế và hồi cứu về mối quan hệ của NMOSD và thai nghén. Cho thấy NMOSD có liên quan đến tăng nguy cơ sẩy thai, và tỷ lệ tái phát NMOSD hàng năm tăng lên trong ba đến sáu tháng đầu của thời kỳ hậu sản.
Nguồn UpToDate 2018
Đọc thêm
Tiêu chuẩn chẩn đoán NMO và NMOSD
Điều trị NMO và NMOSD- cập nhật 2018
Tiếp cân BN mất thị lực 2 bên và liệt tứ chi
Chẩn đoán bệnh nhân giảm thị lực 2 bên
Tin cùng chuyên mục:
Migraine tiền đình (vestibular migraine)
Migraine ở phụ nữ có thai
Cập nhật chẩn đoán và điều trị chóng mặt tư thế kịch phát lành tính (BPPV)
Nguy cơ xuất huyết não tăng lên khi dùng Aspirin liều thấp trong phòng ngừa nguyên phát